
Guo, X.; Qu, Z.; Gao, J. Chem. Phys. Lett., 2018, 691, 91-97.
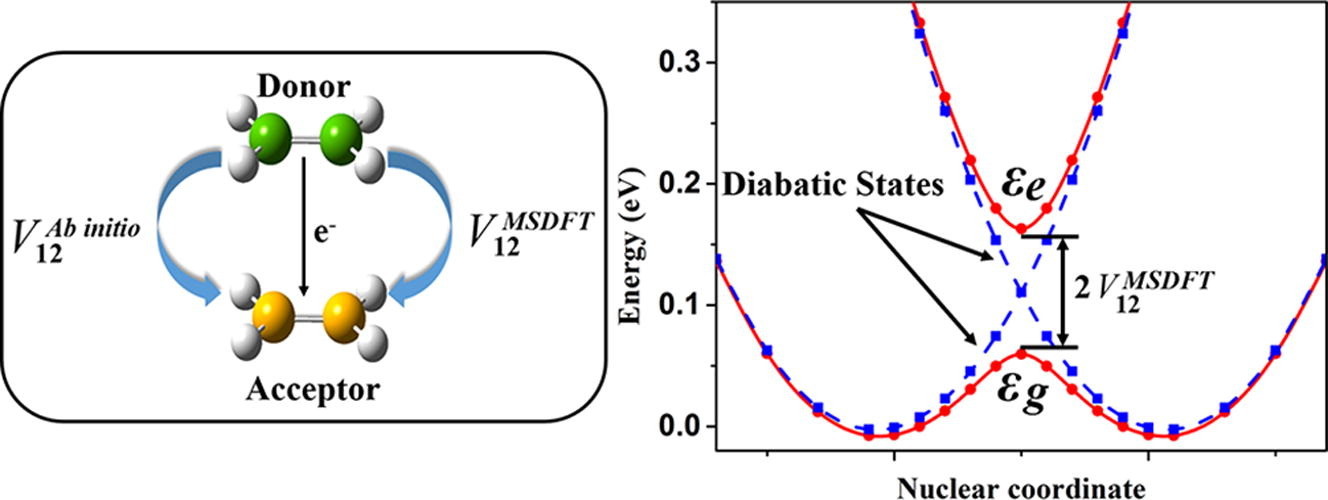
The multi-state density functional theory (MSDFT) provides a convenient way to estimate electronic coupling of charge transfer processes based on a diabatic representation. Its performance has been benchmarked against the HAB11 database with a mean unsigned error (MUE) of 17 meV between MSDFT and ab initio methods. The small difference may be attributed to different representations, diabatic from MSDFT and adiabatic from ab initio calculations. In this discussion, we conclude that MSDFT provides a general and efficient way to estimate the electronic coupling for charge-transfer rate calculations based on the Marcus-Hush model.