
Han, J.; Zhao, R.; Guo, Y.; Qu, Z,; Gao, J.. Molecules, 2022, 27(11), 3466.
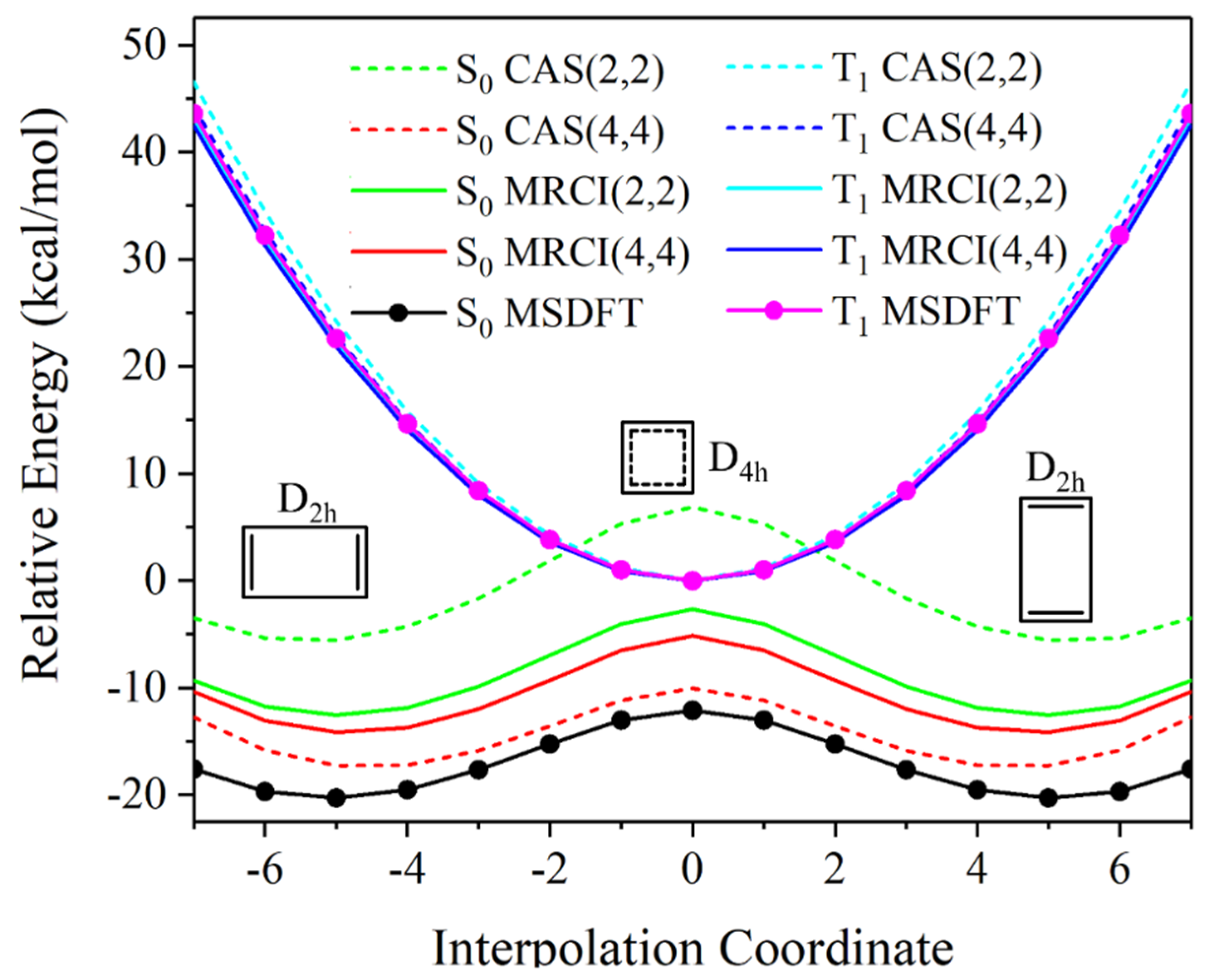
This work explores the electronic structure as well as the reactivity of singlet diradicals, making use of multistate density functional theory (MSDFT). In particular, we show that a minimal active space of two electrons in two orbitals is adequate to treat the relative energies of the singlet and triplet adiabatic ground state as well as the first singlet excited state in many cases. This is plausible because dynamic correlation is included in the first place in the optimization of orbitals in each determinant state via block-localized Kohn–Sham density functional theory. In addition, molecular fragment, i.e., block-localized Kohn–Sham orbitals, are optimized separately for each determinant, providing a variational diabatic representation of valence bond-like states, which are subsequently used in nonorthogonal state interactions (NOSIs). The computational procedure and its performance are illustrated on some prototypical diradical species. It is shown that NOSI calculations in MSDFT can be used to model bond dissociation and hydrogen-atom transfer reactions, employing a minimal number of configuration state functions as the basis states. For p- and s-types of diradicals, the closed-shell diradicals are found to be more reactive than the open-shell ones due to a larger diabatic coupling with the final product state. Such a diabatic representation may be useful to define reaction coordinates for electron transfer, proton transfer and coupled electron and proton transfer reactions in condensed-phase simulations.